Tin tức
Chứng nhận CE cho thiết bị y tế: MDD 93/42/EEC & MDR 2017/745
Trong ngành công nghiệp thiết bị y tế, việc đảm bảo an toàn và hiệu suất là ưu tiên hàng đầu. Đối với các nhà sản xuất đang hướng tới thị trường châu Âu, chứng nhận CE cho thiết bị y tế là một yêu cầu pháp lý bắt buộc và không thể thiếu. Vậy, chứng nhận CE cho thiết bị y tế là gì và nó liên quan như thế nào đến các quy định phức tạp của Liên minh Châu Âu?
Bài viết này sẽ đi sâu giải thích tầm quan trọng của chứng nhận CE cho thiết bị y tế, đặc biệt làm rõ vai trò của Chỉ thị về thiết bị y tế 93/42/EEC và những thay đổi quan trọng khi chuyển đổi sang Quy định thiết bị y tế của EU (MDR) 2017/745, cung cấp cái nhìn toàn diện để bạn tự tin đưa sản phẩm ra thị trường quốc tế.
Chứng nhận CE cho thiết bị y tế là gì?

Để hiểu rõ chứng nhận CE cho thiết bị y tế là gì, chúng ta cần nhìn nhận nó như một bằng chứng pháp lý khẳng định sản phẩm y tế đã đáp ứng các yêu cầu nghiêm ngặt về an toàn và hiệu suất của Liên minh Châu Âu. Đây không chỉ là một con dấu đơn thuần mà là kết quả của một quá trình đánh giá sự phù hợp toàn diện, đảm bảo rằng thiết bị y tế được thiết kế, sản xuất và đưa ra thị trường một cách an toàn nhất cho bệnh nhân và người sử dụng.
Các sản phẩm thiết bị y tế muốn được lưu hành hợp pháp tại EU buộc phải có dấu CE Mark. Điều này có nghĩa là chúng phải tuân thủ các chỉ thị và quy định liên quan của EU. Mục tiêu chính của chứng nhận CE cho thiết bị y tế là bảo vệ sức khỏe và an toàn cộng đồng, đồng thời tạo điều kiện thuận lợi cho việc lưu thông tự do của các thiết bị y tế an toàn và hiệu quả trong thị trường nội địa châu Âu.
Trước đây, nền tảng chính cho chứng nhận CE cho thiết bị y tế là Chỉ thị về thiết bị y tế 93/42/EEC (Medical Devices Directive – MDD). Chỉ thị này đã đặt ra các yêu cầu chung về an toàn và hiệu suất cho một loạt các thiết bị y tế. Tuy nhiên, với sự phát triển của công nghệ và nhận thức về an toàn, Chỉ thị MDD đã được thay thế bằng một quy định mới và toàn diện hơn, đó là Quy định về thiết bị y tế của EU (Medical Device Regulation – MDR) số 2017/745. Sự chuyển đổi này đánh dấu một bước tiến lớn trong việc siết chặt các yêu cầu và quy trình đối với chứng nhận CE cho thiết bị y tế, nhằm nâng cao hơn nữa mức độ an toàn và minh bạch.
Chỉ thị về thiết bị y tế 93/42/EEC (MDD): Nền tảng trước đây
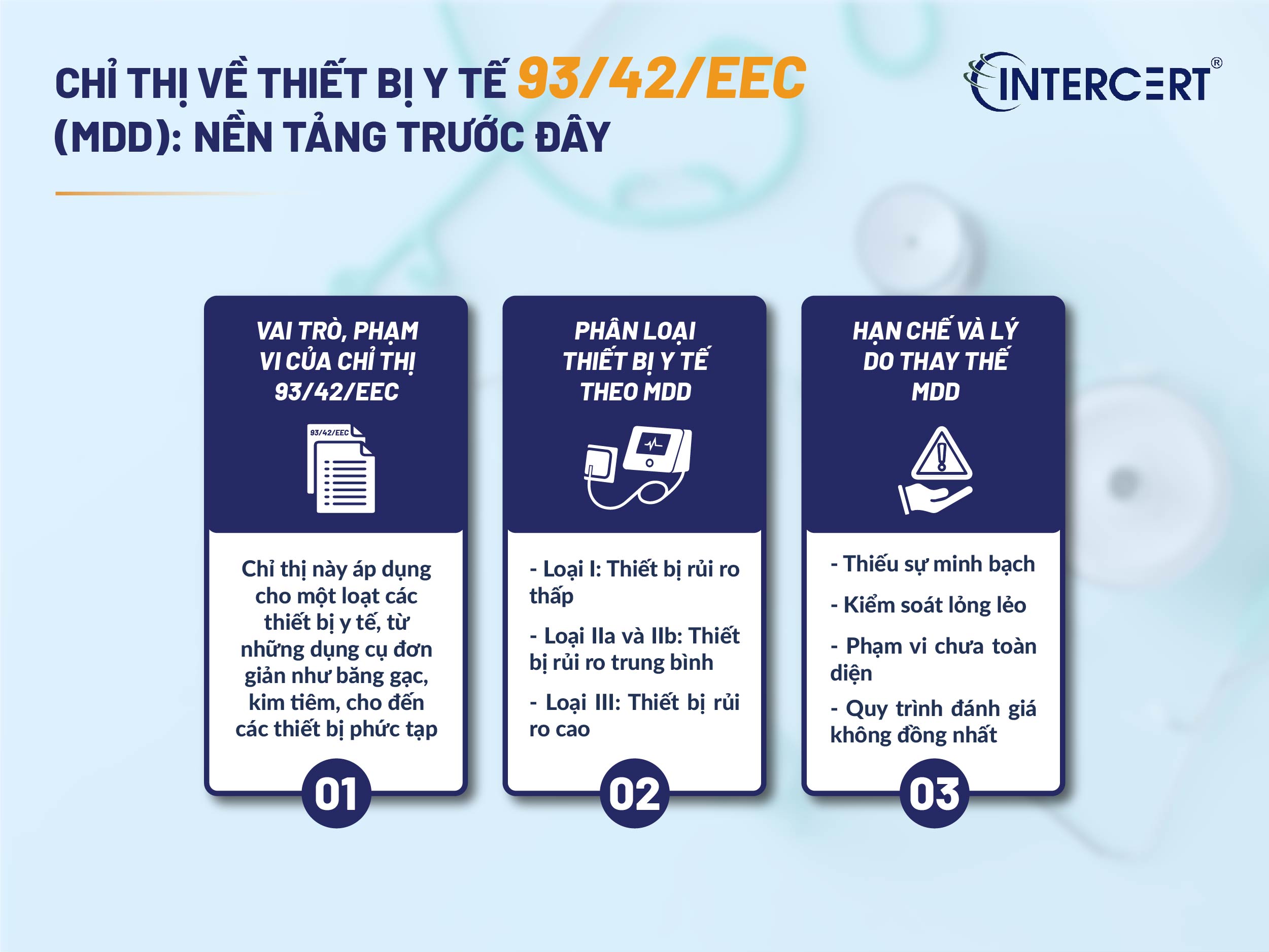
Trong nhiều thập kỷ, Chỉ thị về thiết bị y tế 93/42/EEC (MDD) là văn bản pháp lý chủ chốt quy định các yêu cầu đối với thiết bị y tế tại Liên minh Châu Âu. Việc hiểu rõ MDD là cần thiết để nhìn nhận bức tranh tổng thể về chứng nhận CE cho thiết bị y tế, cũng như nhận biết được những thay đổi quan trọng đã diễn ra.
1. Vai trò và phạm vi của Chỉ thị 93/42/EEC
Chỉ thị về thiết bị y tế 93/42/EEC (MDD) được ban hành nhằm hài hòa các luật quốc gia liên quan đến thiết bị y tế trong EU, đảm bảo rằng tất cả các sản phẩm được đưa ra thị trường đều đáp ứng các yêu cầu cơ bản về an toàn và hiệu suất. Chỉ thị này áp dụng cho một loạt các thiết bị y tế, từ những dụng cụ đơn giản như băng gạc, kim tiêm, cho đến các thiết bị phức tạp hơn như máy MRI, máy thở, thiết bị cấy ghép.
Các sản phẩm tuân thủ Chỉ thị 93/42/EEC (hoặc các tiêu chuẩn hài hòa liên quan) được coi là đáp ứng các yêu cầu của chỉ thị và phải được dán nhãn CE để được phép lưu hành hợp pháp tại EU. Điều này có nghĩa là nhà sản xuất đã thực hiện đánh giá rủi ro, thử nghiệm và lập hồ sơ kỹ thuật để chứng minh rằng thiết bị của họ an toàn và hoạt động như dự định.
2. Phân loại thiết bị y tế theo MDD
MDD phân loại thiết bị y tế thành các nhóm dựa trên mức độ rủi ro tiềm ẩn của chúng, từ đó quy định các quy trình đánh giá sự phù hợp khác nhau:
- Loại I: Thiết bị rủi ro thấp (ví dụ: băng, kính đeo mắt). Hầu hết các thiết bị loại I có thể được tự chứng nhận bởi nhà sản xuất.
- Loại IIa và IIb: Thiết bị rủi ro trung bình (ví dụ: ống tiêm, máy siêu âm). Cần sự tham gia của một Tổ chức được thông báo (Notified Body).
- Loại III: Thiết bị rủi ro cao (ví dụ: thiết bị cấy ghép, thiết bị hỗ trợ sự sống). Yêu cầu sự kiểm soát nghiêm ngặt nhất và sự tham gia bắt buộc của Notified Body.
Việc phân loại đúng là bước quan trọng đầu tiên để xác định quy trình chứng nhận CE cho thiết bị y tế theo MDD.
3. Hạn chế và lý do thay thế MDD
Mặc dù MDD đã phục vụ hiệu quả trong nhiều năm, nhưng sự phát triển nhanh chóng của công nghệ y tế, sự phức tạp ngày càng tăng của các thiết bị và một số sự cố liên quan đến an toàn đã bộc lộ những hạn chế của nó. Một số vấn đề chính bao gồm:
- Thiếu tính minh bạch: Các quy định của MDD đôi khi không đủ rõ ràng, dẫn đến việc giải thích khác nhau giữa các quốc gia thành viên và Notified Body.
- Kiểm soát lỏng lẻo: Quy trình giám sát sau thị trường chưa đủ mạnh mẽ để nhanh chóng phát hiện và xử lý các vấn đề về an toàn.
- Phạm vi chưa toàn diện: MDD chưa bao gồm một số sản phẩm mới hoặc các dịch vụ liên quan đến thiết bị y tế.
- Quy trình đánh giá không đồng nhất: Các Notified Body đôi khi áp dụng các tiêu chuẩn đánh giá khác nhau, tạo ra sự không đồng đều trong mức độ an toàn của sản phẩm.
Những hạn chế này đã thúc đẩy Liên minh Châu Âu xem xét và cuối cùng là ban hành một văn bản pháp lý mới và toàn diện hơn: Quy định về thiết bị y tế (MDR) 2017/745, nhằm tăng cường an toàn cho bệnh nhân và nâng cao độ tin cậy của chứng nhận CE cho thiết bị y tế.
Chuyển đổi từ MDD 93/42/EEC sang Quy định MDR (EU) 2017/745

Một trong những thông tin quan trọng nhất cần nắm về chứng nhận CE cho thiết bị y tế hiện nay là sự thay thế của Chỉ thị về thiết bị y tế 93/42/EEC (MDD) bởi Quy định về thiết bị y tế của EU (MDR) số 2017/745. MDR có hiệu lực từ ngày 26/5/2021, đánh dấu một kỷ nguyên mới cho ngành thiết bị y tế ở châu Âu.
1. Lý do và mục tiêu của MDR
Sự ra đời của MDR không phải là một sự thay đổi nhỏ mà là một cuộc cải cách lớn. MDR được thiết kế để khắc phục những hạn chế của MDD và mang lại một khuôn khổ pháp lý mạnh mẽ hơn, minh bạch hơn và nhất quán hơn cho thiết bị y tế. Các mục tiêu chính của MDR bao gồm:
- Nâng cao mức độ an toàn cho bệnh nhân: Tăng cường các yêu cầu về lâm sàng, đánh giá rủi ro và giám sát sau thị trường.
- Tăng cường tính minh bạch và khả năng truy xuất nguồn gốc: Thiết lập cơ sở dữ liệu thiết bị y tế châu Âu (EUDAMED) để cải thiện việc chia sẻ thông tin và giám sát.
- Cải thiện chức năng của Notified Body: Quy định chặt chẽ hơn về việc chỉ định, giám sát và trách nhiệm của các Notified Body.
- Đảm bảo cạnh tranh công bằng: Áp dụng các quy tắc nhất quán trên toàn EU để tránh sự phân biệt đối xử.
- Thích ứng với sự đổi mới công nghệ: Phạm vi của MDR được mở rộng để bao gồm cả các sản phẩm không có mục đích y tế nhưng có rủi ro tương tự (ví dụ: kính áp tròng màu thẩm mỹ).
2. Những thay đổi chính trong MDR ảnh hưởng đến Chứng nhận CE
MDR mang đến nhiều thay đổi quan trọng tác động trực tiếp đến quy trình chứng nhận CE cho thiết bị y tế:
- Phạm vi áp dụng rộng hơn: MDR bao gồm nhiều loại sản phẩm hơn, bao gồm cả thiết bị y tế không có mục đích y tế dự kiến (như thiết bị làm đẹp có rủi ro cao).
- Quy trình phân loại lại: Một số thiết bị có thể bị phân loại lại lên nhóm rủi ro cao hơn, dẫn đến yêu cầu quy trình đánh giá sự phù hợp chặt chẽ hơn và bắt buộc có sự tham gia của Notified Body.
- Yêu cầu về bằng chứng lâm sàng tăng cường: Nhà sản xuất cần cung cấp bằng chứng lâm sàng mạnh mẽ hơn cho thiết bị của họ, bao gồm dữ liệu từ các nghiên cứu lâm sàng hoặc đánh giá lâm sàng toàn diện hơn.
- Hồ sơ kỹ thuật chi tiết hơn: Các yêu cầu về tài liệu kỹ thuật trở nên chi tiết và nghiêm ngặt hơn, đòi hỏi nhà sản xuất phải cung cấp thông tin minh bạch và toàn diện hơn.
- Hệ thống số nhận dạng thiết bị duy nhất (UDI): MDR yêu cầu mỗi thiết bị phải có một mã UDI để cải thiện khả năng truy xuất nguồn gốc trong chuỗi cung ứng.
- Giám sát sau thị trường (Post-Market Surveillance – PMS) chặt chẽ hơn: Nhà sản xuất có nghĩa vụ thu thập và phân tích dữ liệu về hiệu suất và an toàn của thiết bị sau khi chúng được đưa ra thị trường, và báo cáo bất kỳ sự cố nghiêm trọng nào.
- Người chịu trách nhiệm tuân thủ quy định (PRRC): Yêu cầu nhà sản xuất chỉ định ít nhất một người chịu trách nhiệm về việc tuân thủ các quy định của MDR.
Quy trình đạt chứng nhận CE cho thiết bị y tế theo MDR
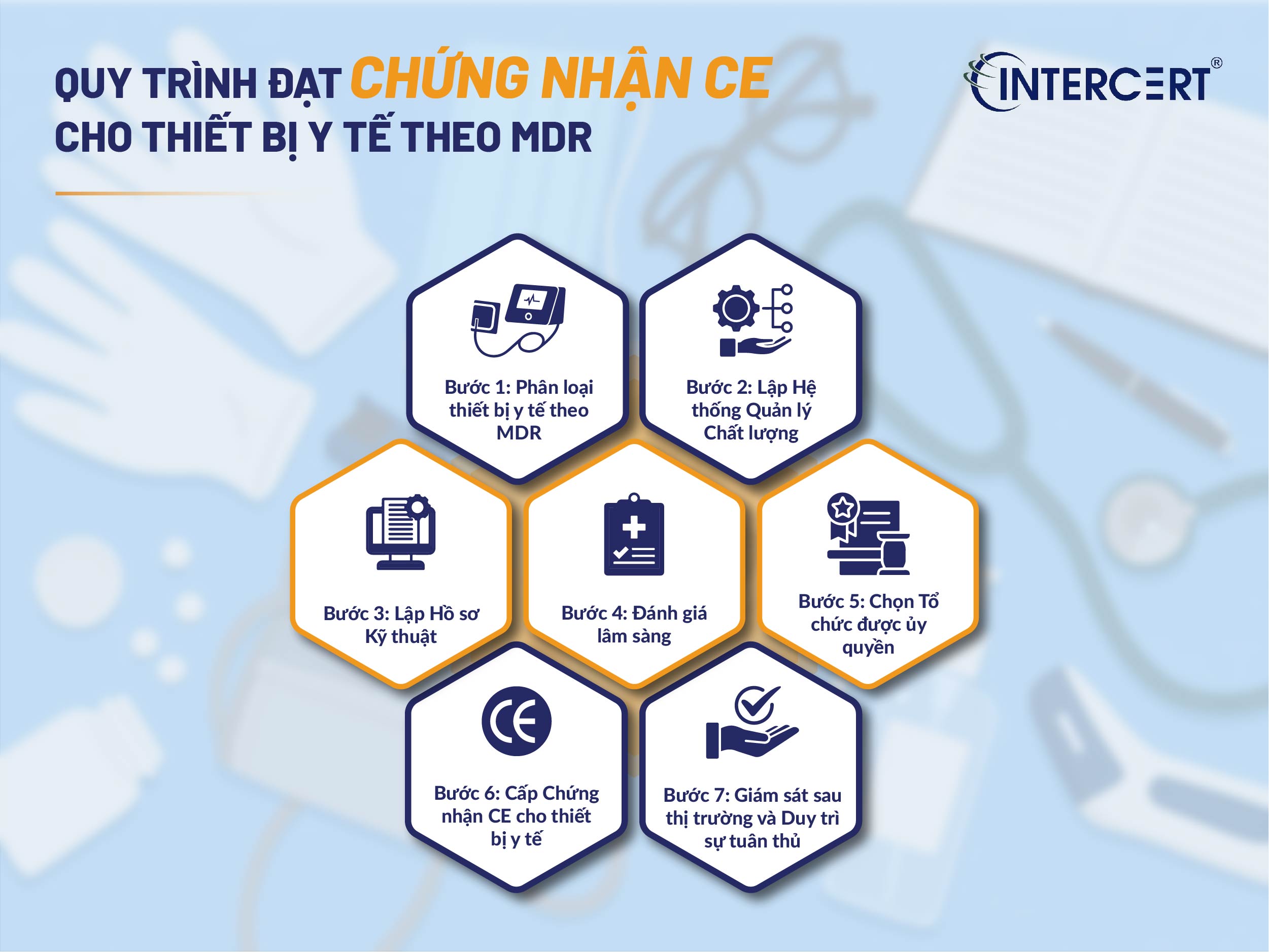
Với sự thay đổi từ MDD sang MDR, quy trình đạt chứng nhận CE cho thiết bị y tế đã trở nên chặt chẽ và phức tạp hơn. Nhà sản xuất cần hiểu rõ các bước để đảm bảo sự tuân thủ.
Bước 1: Phân loại thiết bị y tế theo MDR
Đây là bước đầu tiên và quan trọng nhất, quyết định toàn bộ quy trình đánh giá sự phù hợp. MDR có 22 quy tắc phân loại, chi tiết hơn và có thể dẫn đến việc phân loại lại nhiều thiết bị vào nhóm rủi ro cao hơn so với MDD. Việc phân loại đúng là nền tảng để xác định các yêu cầu tiếp theo.
Bước 2: Lập Hệ thống Quản lý Chất lượng (QMS)
Nhà sản xuất phải thiết lập và duy trì một Hệ thống Quản lý Chất lượng (QMS) toàn diện, tuân thủ các yêu cầu của MDR (thường dựa trên tiêu chuẩn ISO 13485). QMS cần bao trùm toàn bộ vòng đời sản phẩm, từ thiết kế, phát triển, sản xuất, kiểm soát chất lượng đến giám sát sau thị trường.
Bước 3: Lập Hồ sơ Kỹ thuật (Technical Documentation)
Hồ sơ kỹ thuật theo MDR yêu cầu chi tiết và minh bạch hơn nhiều so với MDD. Nó phải cung cấp bằng chứng rõ ràng về sự tuân thủ các yêu cầu chung về an toàn và hiệu suất của thiết bị. Hồ sơ này cần bao gồm:
- Mô tả và thông số kỹ thuật của thiết bị.
- Thông tin về thiết kế và sản xuất.
- Kết quả phân tích rủi ro.
- Bằng chứng lâm sàng (Clinical Evaluation Report – CER).
- Kết quả thử nghiệm (cả thử nghiệm tiền lâm sàng và lâm sàng).
- Nhãn mác và hướng dẫn sử dụng.
- Kế hoạch giám sát sau thị trường (PMS Plan).
Bước 4: Đánh giá lâm sàng (Clinical Evaluation)
MDR nhấn mạnh tầm quan trọng của bằng chứng lâm sàng. Nhà sản xuất phải thực hiện đánh giá lâm sàng toàn diện và liên tục cập nhật, sử dụng dữ liệu lâm sàng để chứng minh sự an toàn và hiệu suất của thiết bị. Đối với các thiết bị rủi ro cao, có thể yêu cầu thử nghiệm lâm sàng (Clinical Investigation) mới.
Bước 5: Lựa chọn Tổ chức được ủy quyền (Notified Body) và Đánh giá sự phù hợp
Đối với hầu hết các loại thiết bị y tế (trừ Loại I không vô trùng/không có chức năng đo lường), sự tham gia của Notified Body là bắt buộc. Notified Body sẽ đánh giá Hồ sơ Kỹ thuật, kiểm tra QMS, và trong một số trường hợp, kiểm tra cả cơ sở sản xuất và quy trình thử nghiệm. Quy trình đánh giá này chặt chẽ hơn nhiều so với MDD.
Bước 6: Cấp Chứng nhận CE cho thiết bị y tế
Sau khi hoàn thành quá trình đánh giá, tổ chức đánh giá sẽ cấp chứng nhận CE cho thiết bị y tế đó. Chứng nhận này sẽ có hiệu lực trong một khoảng thời gian nhất định và cần được gia hạn.
Bước 7: Giám sát sau thị trường và Duy trì sự tuân thủ
MDR yêu cầu nhà sản xuất phải có một hệ thống giám sát sau thị trường (PMS) chủ động và có hệ thống báo cáo các sự cố nghiêm trọng cho cơ quan có thẩm quyền. Việc duy trì sự tuân thủ liên tục, bao gồm cập nhật tài liệu kỹ thuật và QMS, là bắt buộc trong suốt vòng đời của sản phẩm. Không tuân thủ PMS có thể dẫn đến thu hồi chứng nhận CE cho thiết bị y tế.
—————————————————————————————————-
Với sự chuyển đổi từ Chỉ thị về thiết bị y tế 93/42/EEC sang Quy định MDR, các yêu cầu đã trở nên chặt chẽ và toàn diện hơn bao giờ hết. Việc hiểu rõ chứng nhận CE cho thiết bị y tế là gì trong bối cảnh pháp lý hiện tại, cùng với việc chủ động nắm bắt và tuân thủ các quy trình mới của MDR, sẽ là chìa khóa để đảm bảo sản phẩm của bạn không chỉ an toàn và hiệu quả mà còn hợp pháp để tự tin chinh phục thị trường châu Âu đầy tiềm năng. Liên hệ với Intercert Việt Nam qua số Hotline: 0969.555.610 hoặc Email: sales@intercertvietnam.com nếu bạn đang quan tâm tới dịch vụ cấp chứng nhận CE Marking cho thiết bị y tế.





